


Tay-Sachs & Sandhoff Diseases (GM2-gangliosidosis)
Among all of the lysosomal storage disorders, the GM2 gangliosidoses are the most investigated subcategory. They are caused by the impaired degradation of GM2 ganglioside, a reaction which is normally catalyzed by the β-hexosaminidase A (HexA) enzyme. Hex A is a dimer of two proteins subunits, α and β, encoded by the HEXA and HEXB genes. Mutations of these two genes will lead to the accumulation of the GM2-ganglioside within the lysosomes, as observed in both Tay-Sachs (http://www.omim.org/entry/272800) and Sandhoff (http://www.omim.org/entry/268800 ) disease.
In Tay-Sachs disease (TSD), the lysosomal engulfment is confined to neurons and results in a massive apoptosis and a subsequent reactive gliosis. Such a precipitous neurodegeneration primarily affects motor and spinocerebellar functions.
Although more common in certain ethnic groups, Tay-Sachs is a rare disease itself, an incidence of one in 320,000 births in the general population. Depending on the age of disease onset and the enzymatic activity, Tay-Sachs can be further classified into:
- Infantile form: the symptoms begin at 3-5 months of age and consist of motor and regression of previously acquired motor skill, which becomes rapidly fatal after 8-10 months of age. By this time the ophthalmoscopic examination could easily reveal macular pallor and prominence of the fovea centralis: the 'cherry-red spot' sign which, although non-specific, is a common finding in TSD.
- Juvenile form: milder, the first symptoms are observed in children between two and ten years of age and mainly consists of cognitive and motor deterioration, as well as dysarthria, dysphagia and spasticity.
- Adult form: the rarest one, non-fatal and usually misdiagnosed with schizophrenia-like psychoses.
In the clinical setting, TSD can be only addressed with supportive care and management of the intervening problems. Our lab, however, focuses on pre-clinical development of AAV intracranial gene therapy to treat TSD and GM2 gangliosidoses in general. We have shown that direct injection into specific regions of the brain can result in an "enzyme producing center", in which therapeutic enzyme can be expressed from an AAV, secreted and distributed throughout the rest of the brain.
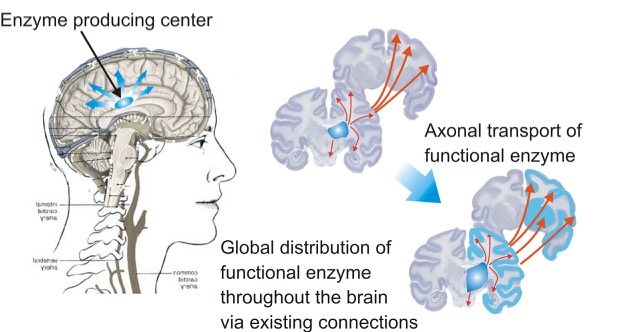
We have had much therapeutic success in treating both small (mouse) and large animal (cat and sheep) models of GM2 gangliosidoses using intracranial gene therapy. We inject an equal mixture of AAVrh8 vectors, which encode for species-specific HEXA or HEXB genes. We have tested six vector designs with a range of expression levels to determine safety and efficacy. After screening for toxicity in mice and primates, as well as additional therapeutic efficacy studies in Sandhoff mice, we have chosen a final vector formulation and final IND-enabling studies in primates and Sandhoff mice are underway.