

Integrated Strategies to Elucidate Molecular Recognition
I. Elucidating Molecular Recognition: Parallel MD Simulations
II. Integrating and Digesting Data: Machine Learning
I. Elucidating Molecular Recognition: Parallel MD Simulations
Molecular recognition is inherently a dynamic event, as biomolecules sample an ensemble of conformations around their native states. Molecular dynamics (MD) simulations enable assessing this flexibility for protein-ligand complexes. In the past decade the Schiffer lab has developed a strategy of parallel Molecular Dynamics simulations we define as pMD to collectively analyze a series of MD simulations of similar yet distinct molecular complexes to decipher conformational and dynamic differences responsible for changes in molecular recognition. We perform pMD simulations on complexes with varying protein sequence and ligand identity to unravel structural and dynamic properties that underlie coupled changes in molecular recognition.
We have applied this pMD strategy to protein-ligand complexes for series of natural substrates, inhibitors, and protease mutations. Combined with experimental data on strength of binding, we determine which physical properties or molecular interactions are key to molecular recognition for a given system. These changes dictate not only the interdependency of molecular recognition or specificity but also inform inhibitor design. Using this approach we are addressing shortcomings in traditional structure based drug design (SBDD), by incorporating into drug design:
- Dynamics (flexibility) of protein–ligand complex
- Impact of mutations remote from the active site
- Impact of coupled changes within a ligand
- Interdependency between sub-sites (or binding grooves) within the active site
II. Integrating and Digesting Data: Machine Learning
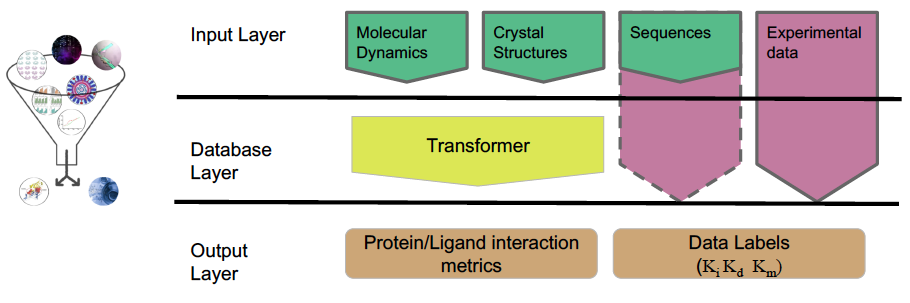
While we mainly use molecular information derived from pMD simulations, this approach can also incorporate experimental and computational data from various techniques. Integrating these input data, we construct physical "fingerprints" of protein–ligand complexes and use machine learning to extract predictors of molecular recognition and deduce key specific alterations.
Relevant Publications
Hydration Structure and Dynamics of Inhibitor-Bound HIV-1 Protease.
Leidner F, Kurt Yilmaz N, Paulsen J, Muller YA, Schiffer CA.
J Chem Theory Comput. 2018 May 8;14(5):2784-2796. doi: 10.1021/acs.jctc.8b00097. Epub 2018 Apr 18.
Interdependence of Inhibitor Recognition in HIV-1 Protease.
Paulsen JL, Leidner F, Ragland DA, Kurt Yilmaz N, Schiffer CA.
J Chem Theory Comput. 2017 May 9;13(5):2300-2309. doi: 10.1021/acs.jctc.6b01262. Epub 2017 Apr 11.