

Research
Early studies on phage recombination functions
Early in my career, I studied components of recombination systems from bacteriophage lambda and phage P22. These studies included an early description of the single-stranded DNA annealing (SSDA) function of phage P22 called Erf (for essential recombination function). It is a functional analog of the well-known λ Beta annealase of the λ Red system, and promotes annealing of two complementary single-stranded DNAs in vitro. The λ Beta protein can substitute for Erf during a P22 phage infection in Salmonella typhimurium, and has a ring like structure with C-terminal protrusions as seen under an electron microscope (like λ Beta). Its domain structure has been analyzed both biochemically and genetically (1).
In addition, other recombination-promoting phage functions were an interest of mine at that time, including phage-encoded modifiers of the host RecBCD enzyme (aka ExoV). While RecBCD is a major component of the main route of homologous recombination in E. coli and S. typhimurium, it is also a powerful ATP-dependent exonuclease. Recombination via RecBCD is dependent on the enzyme’s encounter with the hotspot initiator Chi sequences (GCTGGTGG), which alters the enzymatic properties of RecBCD such that the enzyme generates 3’ ssDNA recombinogenic tails at dsDNA ends. However, both phage λ and P22 do not have Chi sites and thus cannot take advantage of the recombinogenic potential of RecBCD, and instead, are subject to the destructive action of RecBCD’s dsDNA exonuclease. To counteract the action of RecBCD, both phage λ and P22 encode anti-RecBCD modifiers, which both protect their chromosomes for exonucleolytic attack and allow their recombination systems to act unimpeded. However, each anti-RecBCD function is different. While the λ Gam protein binds to RecBCD and prevents the enzyme from binding dsDNA ends (2, 3), the anti-RecBCD function from phage P22 (called Abc) binds to the RecC subunit, inhibits Chi activity, and hijacks the exonuclease activity of RecBCD to promote phage recombination. Phage P22 does not have an λ-like exonuclease; instead, it uses a modified RecBCD exonuclease function to promote phage recombination (4-6). The Gam protein plays an important role in recombineering by protecting linear dsDNA substrates following electroporation into E. coli, giving time and opportunity for the λ Red system to work efficiently.
Discovery of gene replacement in E. coli using the λ phage Red recombination system.
E. coli gene replacement technologies in the 1990s consisted of two major avenues. One was to transform a non-replicating plasmid that contained a mutant version of the target gene. This was often a very time-consuming process that relied on endogenous recombination systems, occurred at low frequencies, and required a second counter-selection step to resolve the co-integrant. The second method was the use of recombination–proficient strains of E. coli (sbcA, recBC sbcB15, or recD) that increased the frequency of transformation with linear dsDNA species, albeit to a low and variably efficient rate. It became evident at the time that the genotypes of these recombination–proficient strains were similar: they all resulted in inactivation of the host RecBCD dsDNA exonuclease, and induced an alternate homologous recombination pathway. These were both properties of the phage lambda Red pathway that I had been studying at the time. Thus, I tested whether λ Red and Gam could be used to promote recombination of linear dsDNAs with the bacterial chromosome, by producing these functions from a plasmid or by substituting the chromosomal recBCD region with the Plac-red-gam functions. To my surprise, linear dsDNA transformation frequencies were 100 times higher with λ Red and Gam expression relative to recD and recBC sbcB15 strains being used for gene replacement at the time (7). This was the first demonstration of the λ Red gene replacement methodology. I shared my results with both the Court and Wanner labs, who later showed that such recombination frequencies only requires ~ 50 bp of flanking homologies, a result I had discovered as well, by expressing the phage functions from a stronger promoter (Ptac, instead of Plac). The use of such short homologies had already been demonstrated in the Stewart lab, using the RecET recombination system from the rac prophage. The process became known as “recombineering” and has revolutionized the methods of bacterial genetic engineering. Some of my contributions to this field are highlighted in primary articles and reviews listed below (8-13).
Genetic engineering of pathogenic bacteria
In 2003, I was able to demonstrate the successful use of the Red system in enterohemorrhagic E. coli (EHEC), as well as enteropathogenic E. coli (EPEC) (13). The λ Red-producing plasmid for performing recombineering in these pathogens, pKM208, has been cited in numerous articles and is available form the addgene.com plasmid repository website. Other work representing recombineering of EHEC strains from my lab are listed below (14-17). In 2007, my research took a turn toward the development of genetic engineering strategies for Mycobacterium tuberculosis, highlighted by an extensive and continuing collaboration with colleague Christopher Sassetti here in the Microbiology department. Studies toward this end and current work regarding DNA mismatch repair mechanisms in mycobacteria can be found below.
1) Murphy KC, Casey L, Yannoutsos N, Poteete AR, Hendrix RW. Localization of a DNA-binding determinant in the bacteriophage P22 Erf protein. J Mol Biol. 1987 Mar 5;194(1):105-17. doi: 10.1016/0022-2836(87)90719-4. PubMed PMID: 3612797.
2) Murphy KC. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J Bacteriol. 1991 Sep;173(18):5808-21. doi: 10.1128/jb.173.18.5808-5821.1991. PubMed PMID: 1653221; PubMed Central PMCID: PMC208314.
3) Murphy KC. The lambda Gam protein inhibits RecBCD binding to dsDNA ends. J Mol Biol. 2007 Aug 3;371(1):19-24. doi: 10.1016/j.jmb.2007.05.085. Epub 2007 Jun 2. PubMed PMID: 17583735.
4) Murphy KC. Biochemical characterization of P22 phage-modified Escherichia coli RecBCD enzyme. J Biol Chem. 1994 Sep 9;269(36):22507-16. PubMed PMID: 8077199. select
5) Murphy KC, Lewis LJ. Properties of Escherichia coli expressing bacteriophage P22 Abc (anti-RecBCD) proteins, including inhibition of Chi activity. J Bacteriol. 1993 Mar;175(6):1756-66. doi: 10.1128/jb.175.6.1756-1766.1993. PubMed PMID: 8383665; PubMed Central PMCID: PMC203970.
6) Murphy KC. Bacteriophage P22 Abc2 protein binds to RecC increases the 5' strand nicking activity of RecBCD and together with lambda bet, promotes Chi-independent recombination. J Mol Biol. 2000 Feb 18;296(2):385-401. doi: 10.1006/jmbi.1999.3486. PubMed PMID: 10669596.
7) Murphy KC. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol. 1998 Apr;180(8):2063-71. doi: 10.1128/JB.180.8.2063-2071.1998. PubMed PMID: 9555887; PubMed Central PMCID: PMC107131.
8) Murphy KC, Campellone KG, Poteete AR. PCR-mediated gene replacement in Escherichia coli. Gene. 2000 Apr 4;246(1-2):321-30. doi: 10.1016/s0378-1119(00)00071-8. PubMed PMID: 10767554.
9) Murphy KC, Marinus MG. RecA-independent single-stranded DNA oligonucleotide-mediated mutagenesis. F1000 Biol Rep. 2010 Jul 22;2:56. doi: 10.3410/B2-56. PubMed PMID: 20711416; PubMed Central PMCID: PMC2920528.
10) Murphy KC. Targeted chromosomal gene knockout using PCR fragments. Methods Mol Biol. 2011;765:27-42. doi: 10.1007/978-1-61779-197-0_2. PubMed PMID: 21815084.
11) Murphy KC. Phage recombinases and their applications. Adv Virus Res. 2012;83:367-414. doi: 10.1016/B978-0-12-394438-2.00008-6. Review. PubMed PMID: 22748814.
12) Murphy KC. λ Recombination and Recombineering. EcoSal Plus. 2016 May;7(1). doi: 10.1128/ecosalplus.ESP-0011-2015. Review. PubMed PMID: 27223821.
13) Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol. 2003 Dec 13;4:11. doi: 10.1186/1471-2199-4-11. PubMed PMID: 14672541; PubMed Central PMCID: PMC317293.
14) Savage PJ, Leong JM, Murphy KC. Rapid allelic exchange in enterohemorrhagic Escherichia coli (EHEC) and other E. coli using lambda red recombination. Curr Protoc Microbiol. 2006 Jan;Chapter 5:Unit5A.2. doi: 10.1002/9780471729259.mc05a02s00. PubMed PMID: 18770591.
15)Campellone KG, Roe AJ, Løbner-Olesen A, Murphy KC, Magoun L, Brady MJ, Donohue-Rolfe A, Tzipori S, Gally DL, Leong JM, Marinus MG. Increased adherence and actin pedestal formation by dam-deficient enterohaemorrhagic Escherichia coli O157:H7. Mol Microbiol. 2007 Mar;63(5):1468-81. doi: 10.1111/j.1365-2958.2007.05602.x. PubMed PMID: 17302821; NIHMSID:NIHMS228375.
16) Murphy KC, Ritchie JM, Waldor MK, Løbner-Olesen A, Marinus MG. Dam methyltransferase is required for stable lysogeny of the Shiga toxin (Stx2)-encoding bacteriophage 933W of enterohemorrhagic Escherichia coli O157:H7. J Bacteriol. 2008 Jan;190(1):438-41. doi: 10.1128/JB.01373-
17) Carone BR, Xu T, Murphy KC, Marinus MG. High incidence of multiple antibiotic resistant cells in cultures of in enterohemorrhagic Escherichia coli O157:H7. Mutat Res. 2014 Jan;759:1-8. doi: 10.1016/j.mrfmmm.2013.11.008. Epub 2013 Dec 18. PubMed PMID: 24361397; PubMed Central PMCID: PMC3913999.
Genetic engineering of M. tuberculosis using ORBIT
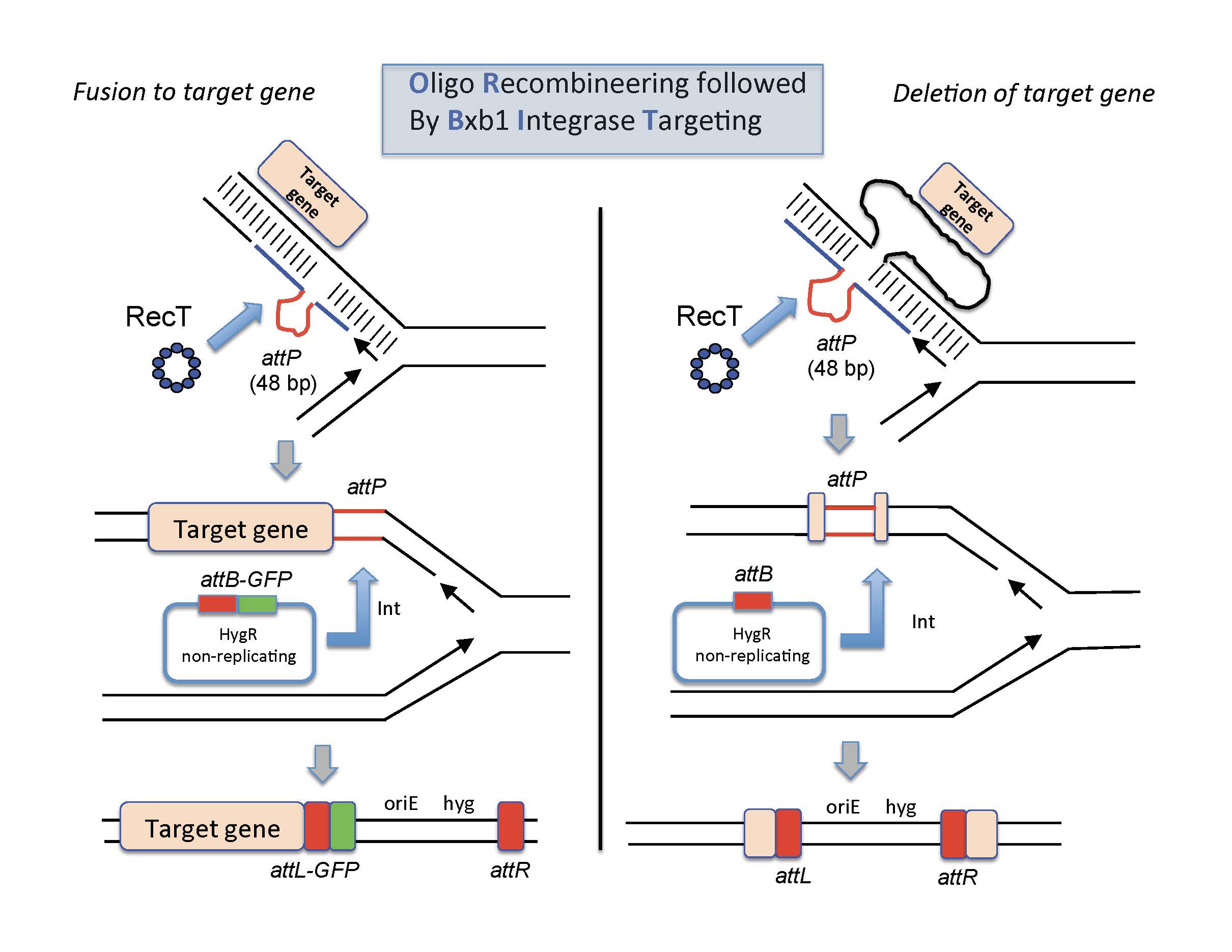
ORBIT-promoted gene alteration. The ORBIT process is initiated at the replication fork. An oligomer containing a single-stranded version of the Bxb1attP site (top pictures, red lines) is coelectroporated with an attB-containing nonreplicating plasmid into a mycobacterial host cell expressing both RecT annealase and Bxb1 Integrase. RecT promotes annealing of the oligonucleotide to the lagging strand template. Following DNA replication through this region, an attP site is formed in the chromosome (middle pictures). In the same outgrowth period, Bxb1 Integrase promotes site-specific insertion of the plasmid into the chromosome (attB x attP). (Left side) The oligonucleotide is designed such that attP is inserted just before the stop codon. The integration event fuses the GFP tag in-frame to the 3’end of the target gene (with an attL site in frame between them); the recombinant is selected for by HygR. (Right side) The oligonucleotide is designed such that attP replaces the target gene and the plasmid integration event allows hygromycin resistance to be used to select for the knockout.
ORBIT-generated insertions and deletions in M. tuberculosis. Target gene deletions and tags were placed at a variety of positions in the chromosomes of M. tuberculosis. In most cases, the oligos contained an attP site flanked by 70 bases of target homology. Insertions (either DAS+4, GFP or His-Flag) are shown in Red and deletions are shown in blue.