


Alpha-1-Antitrypsin Deficiency
Alpha-1 antitrypsin (AAT) is one of the primary circulating serum antiproteases in humans. It inhibits a variety of serine proteinases, with neutrophil elastase being one of the most physiologically important, as well as inhibiting a number of metallo-proteinases and other pro-inflammatory and pro-apoptotic molecules. AAT is normally produced within hepatocytes and macrophages, where hepatocyte-derived AAT forms the bulk of the physiologic reserve of AAT.
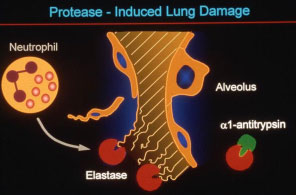
Approximately 4% of the North American and Northern European populations possess at least one copy of a mutant allele, known as PI*Z, which results from a single amino acid substitution of Lys for Glu at position 342. In the homozygous state, this mutation leads to severe deficiency of AAT, and can result in two distinct pathologic states: a lung disease which is primarily due to the loss of antiprotease function, and a liver disease (present to a significant degree in only 10% of patients) which is due to a toxic gain of function of the Z-AAT mutant protein.
The mutation of Z-AAT leads to the loss of a crucial salt-bridge in a beta sheet region of the protein, allowing for the insertion of the reactive loop of a neighboring AAT molecule in between the two beta sheets. The structure of the resultant very stable loop-sheet polymers has been solved and provides insight into how the mutant may accumulate in a misfolded configuration. This accumulation within hepatocytes consistently results in a state of serum deficiency due to inefficient secretion. In individuals affected by AAT liver disease, it also triggers a cascade of aberrant signals within the hepatocyte, most likely the result of an unfolded protein response. However, the downstream details remain unclear, as does the reason for the disparity between those Z-homozygotes who develop liver disease and those who do not. Regardless of the precise mechanism by which Z-AAT conformational changes lead to liver disease, there is general consensus that molecular therapies for AAT liver disease should act by down-regulating Z-AAT.
Our group has developed two different investigational clinical gene therapy products for gene augmentation of AAT as a potential therapy for the lung disease (rAAV2-AAT and rAAV1-AAT) (1, 4), and we have a third vector in development that is much more efficient for delivery of wild-type (M) AAT to hepatocytes (rAAV8-AAT). Given the need for down-regulation of Z-AAT, we have also begun to examine a number of innovative approaches to long-term expression of therapeutic RNAs using the recombinant adeno-associated virus (rAAV) platform, including spliceosome-mediated RNA trans-splicing (SMaRT), and rAAV-mediated in vivo delivery of RNAi in Z-AAT transgenic mice, a project which was recently published from our laboratory. The constructs in that paper utilized a pol III promoter (U6) to express short hairpin RNAs (shRNAs) targeting three different sites on the AAT molecule, but were not specific to the mutant allele. While it is feasible to direct silencing agents to the liver to decrease Z-AAT expression, while directing gene augmentation to other sites, the hepatocyte is most likely the optimal target for augmentation as well.